- HADDOCK server status for docking run E2A-HPR
- Status: FINISHED
- Your HADDOCK run has successfully completed. The complete run can be downloaded as a gzipped tar file here. The file containing your docking parameters is here.
- Please cite the following paper in your work:
G.C.P van Zundert, J.P.G.L.M. Rodrigues, M. Trellet, C. Schmitz, P.L. Kastritis, E. Karaca, A.S.J. Melquiond, M. van Dijk, S.J. de Vries and (2016). "The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes."
J. Mol. Biol., 428, 720-725 (2015).- Questions / feedback ? ask.bioexcel.eu
- Please also consider giving us some feedback by filling our online survey.
- Summary
- HADDOCK clustered 193 structures in 6 cluster(s), which represents 96.5 % of the water-refined models HADDOCK generated. Note that currently the maximum number of models considered for clustering is 200.
- The statistics of the top 10 clusters are shown below. The top cluster is the most reliable according to HADDOCK. Its Z-score indicates how many standard deviations from the average this cluster is located in terms of score (the more negative the better).
- A graphical representation of the results is also provided at the bottom of the page.
- Please cite the following paper in your work:
- Cluster 1
- View the docking solutions in a Jmol structure viewer. Your browser must be Java enabled:
| HADDOCK score | -137.4 +/- 2.7 |
| Cluster size | 114 |
| RMSD from the overall lowest-energy structure | 0.7 +/- 0.6 |
| Van der Waals energy | -40.7 +/- 6.5 |
| Electrostatic energy | -440.8 +/- 82.6 |
| Desolvation energy | -9.6 +/- 12.4 |
| Restraints violation energy | 11.2 +/- 2.23 |
| Buried Surface Area | 1556.0 +/- 31.9 |
| Z-Score | -1.8 |
| Nr 1 best structure | Download structure |
| Nr 2 best structure | Download structure |
| Nr 3 best structure | Download structure |
| Nr 4 best structure | Download structure |
- Cluster 2
- View the docking solutions in a Jmol structure viewer. Your browser must be Java enabled:
| HADDOCK score | -112.8 +/- 2.3 |
| Cluster size | 33 |
| RMSD from the overall lowest-energy structure | 10.9 +/- 0.1 |
| Van der Waals energy | -40.5 +/- 2.5 |
| Electrostatic energy | -311.2 +/- 22.8 |
| Desolvation energy | -11.3 +/- 3.8 |
| Restraints violation energy | 11.8 +/- 3.29 |
| Buried Surface Area | 1597.7 +/- 60.2 |
| Z-Score | -0.4 |
| Nr 1 best structure | Download structure |
| Nr 2 best structure | Download structure |
| Nr 3 best structure | Download structure |
| Nr 4 best structure | Download structure |
- Cluster 3
- View the docking solutions in a Jmol structure viewer. Your browser must be Java enabled:
| HADDOCK score | -111.8 +/- 4.4 |
| Cluster size | 19 |
| RMSD from the overall lowest-energy structure | 8.8 +/- 0.7 |
| Van der Waals energy | -38.4 +/- 3.6 |
| Electrostatic energy | -290.9 +/- 53.9 |
| Desolvation energy | -16.2 +/- 6.7 |
| Restraints violation energy | 9.6 +/- 7.69 |
| Buried Surface Area | 1462.3 +/- 81.2 |
| Z-Score | -0.3 |
| Nr 1 best structure | Download structure |
| Nr 2 best structure | Download structure |
| Nr 3 best structure | Download structure |
| Nr 4 best structure | Download structure |
- Cluster 6
- View the docking solutions in a Jmol structure viewer. Your browser must be Java enabled:
| HADDOCK score | -102.2 +/- 7.8 |
| Cluster size | 5 |
| RMSD from the overall lowest-energy structure | 7.4 +/- 1.3 |
| Van der Waals energy | -35.1 +/- 2.7 |
| Electrostatic energy | -307.7 +/- 18.0 |
| Desolvation energy | -8.7 +/- 2.3 |
| Restraints violation energy | 30.6 +/- 28.43 |
| Buried Surface Area | 1357.4 +/- 34.0 |
| Z-Score | 0.2 |
| Nr 1 best structure | Download structure |
| Nr 2 best structure | Download structure |
| Nr 3 best structure | Download structure |
| Nr 4 best structure | Download structure |
- Cluster 4
- View the docking solutions in a Jmol structure viewer. Your browser must be Java enabled:
| HADDOCK score | -90.6 +/- 3.6 |
| Cluster size | 16 |
| RMSD from the overall lowest-energy structure | 6.0 +/- 0.4 |
| Van der Waals energy | -26.9 +/- 2.1 |
| Electrostatic energy | -290.1 +/- 18.8 |
| Desolvation energy | -9.6 +/- 4.4 |
| Restraints violation energy | 39.2 +/- 36.29 |
| Buried Surface Area | 1220.9 +/- 96.9 |
| Z-Score | 0.9 |
| Nr 1 best structure | Download structure |
| Nr 2 best structure | Download structure |
| Nr 3 best structure | Download structure |
| Nr 4 best structure | Download structure |
- Cluster 5
- View the docking solutions in a Jmol structure viewer. Your browser must be Java enabled:
| HADDOCK score | -82.8 +/- 12.5 |
| Cluster size | 6 |
| RMSD from the overall lowest-energy structure | 7.9 +/- 1.7 |
| Van der Waals energy | -35.8 +/- 4.1 |
| Electrostatic energy | -186.5 +/- 44.7 |
| Desolvation energy | -13.0 +/- 3.9 |
| Restraints violation energy | 33.9 +/- 15.09 |
| Buried Surface Area | 1291.2 +/- 102.8 |
| Z-Score | 1.3 |
| Nr 1 best structure | Download structure |
| Nr 2 best structure | Download structure |
| Nr 3 best structure | Download structure |
| Nr 4 best structure | Download structure |
- Results analysis
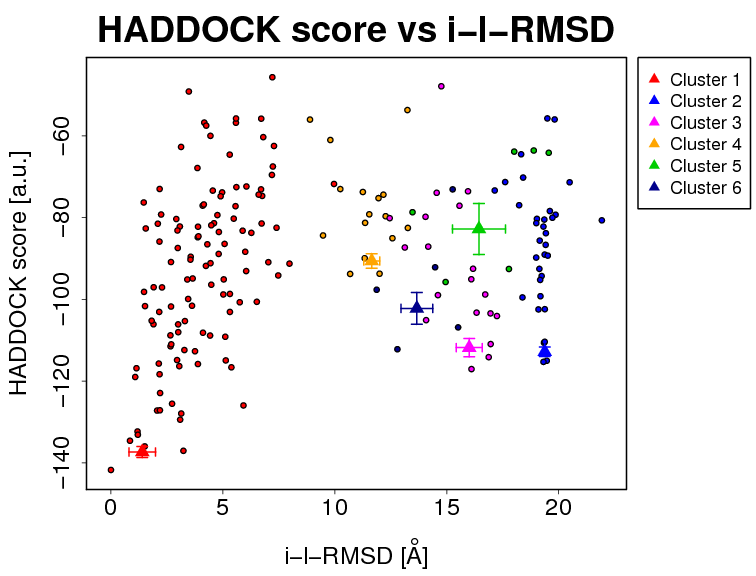
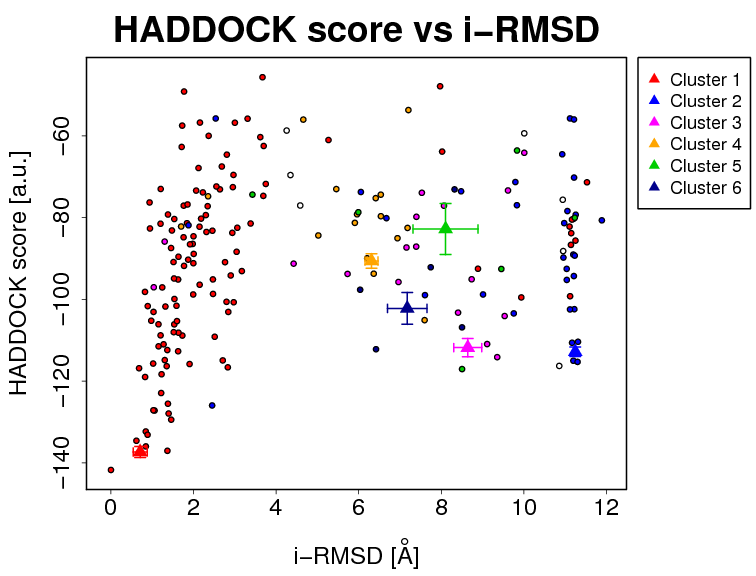
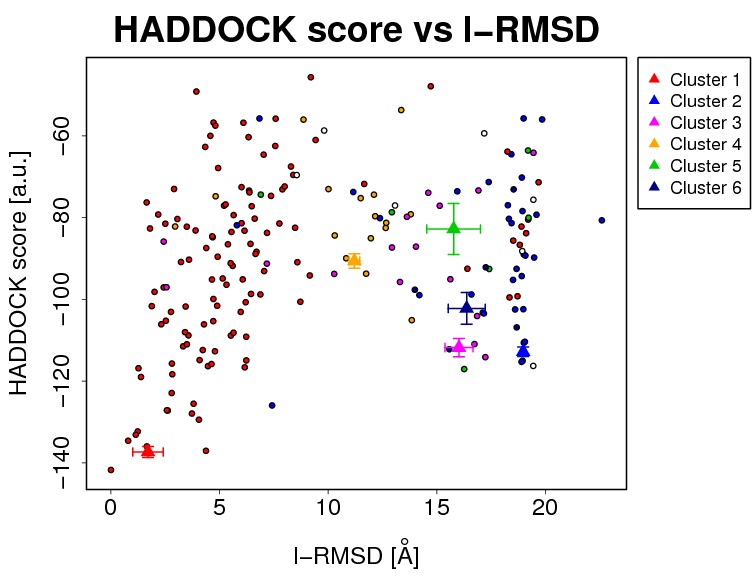
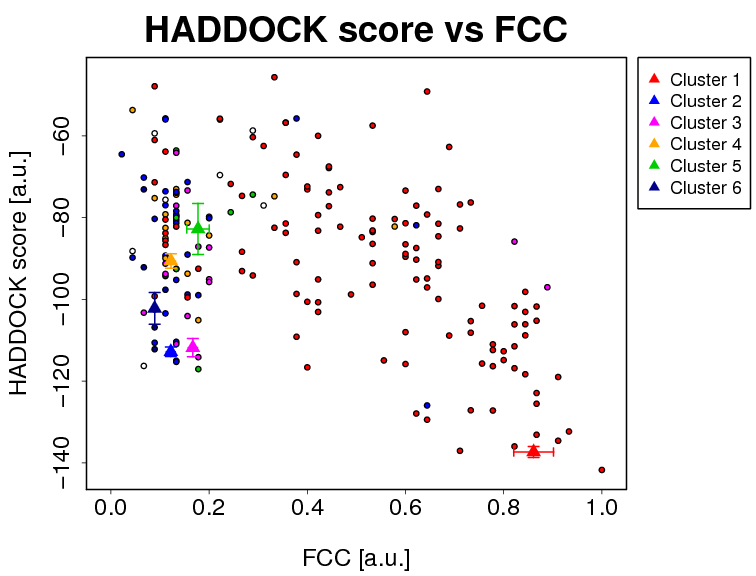
- The results and graphics presented below are based on water-refined models generated by HADDOCK. The clusters (indicated in color in the graphs) are calculated based on the interface-ligand RMSDs calculated by HADDOCK, with the interface defined automatically based on all observed contacts. The various structural analysis (FCC, i-RMSD and l-RMSD) are made with respect to the best HADDOCK model (the one with the lowest HADDOCK score).
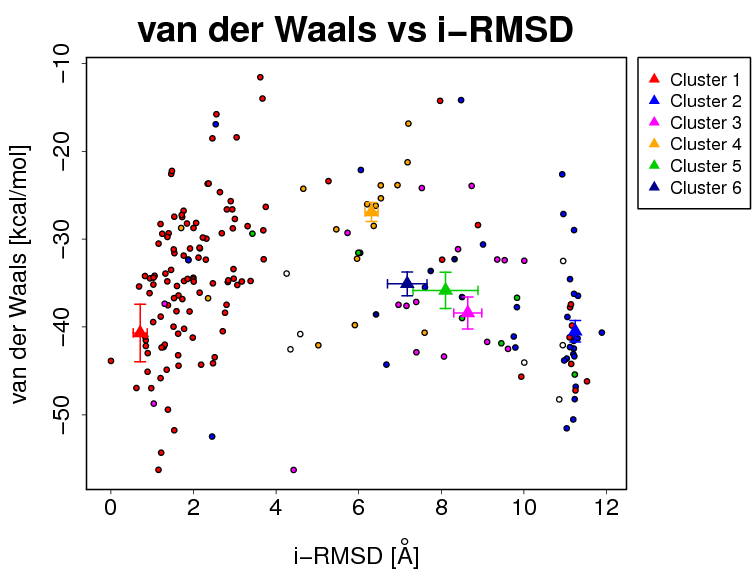
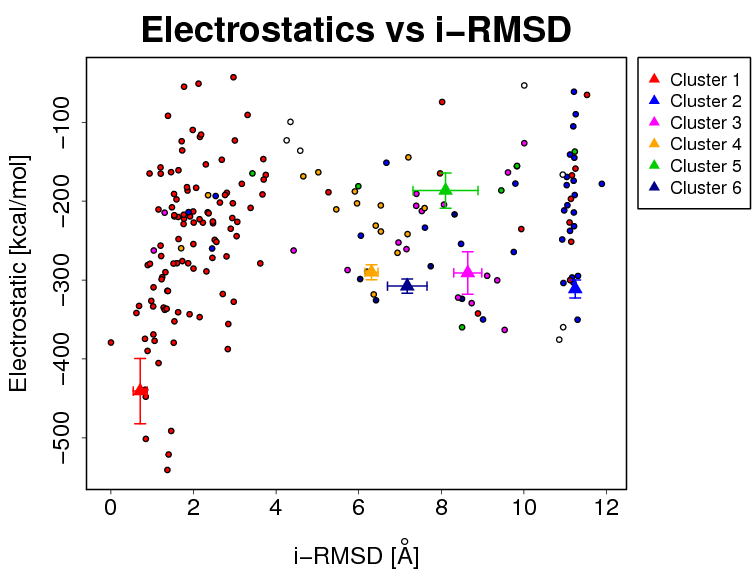
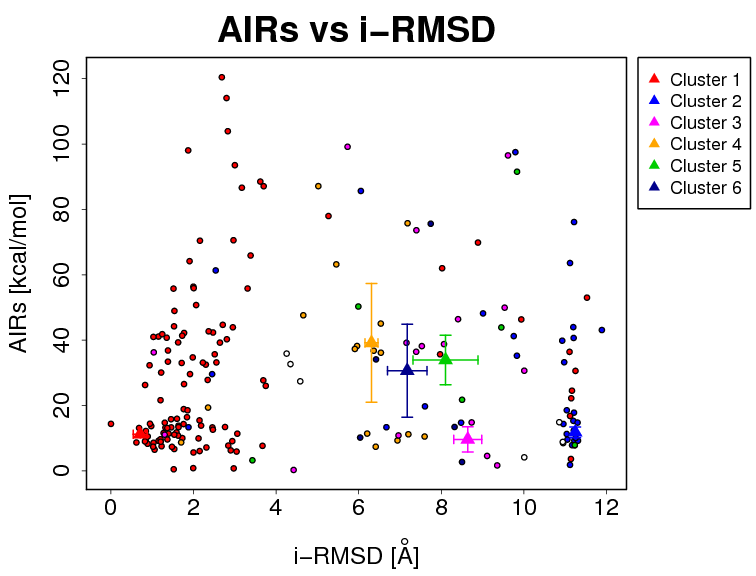
- Supplementary information:
- i-RMSD -> interface-RMSD calculated on the backbone (CA,C,N,O,P) atoms of all residues involved in intermolecular contact using a 10Å cutoff
l-RMSD -> ligand-RMSD calculated on the backbone atoms (CA,C,N,O,P) of all (N>1) molecules after fitting on the backbone atoms of the first (N=1) molecule
FCC -> Fraction of common contacts. The intermolecular contacts are defined based on the best HADDOCK model using a 5Å cutoff (see Rodrigues et al, Proteins 2012)
a.u. -> Arbitrary Units
The cluster averages and standard deviations are indicated by colored dots with associated error bars. The average values are calculated on the best 4 structures of each clusters (based on the HADDOCK score).